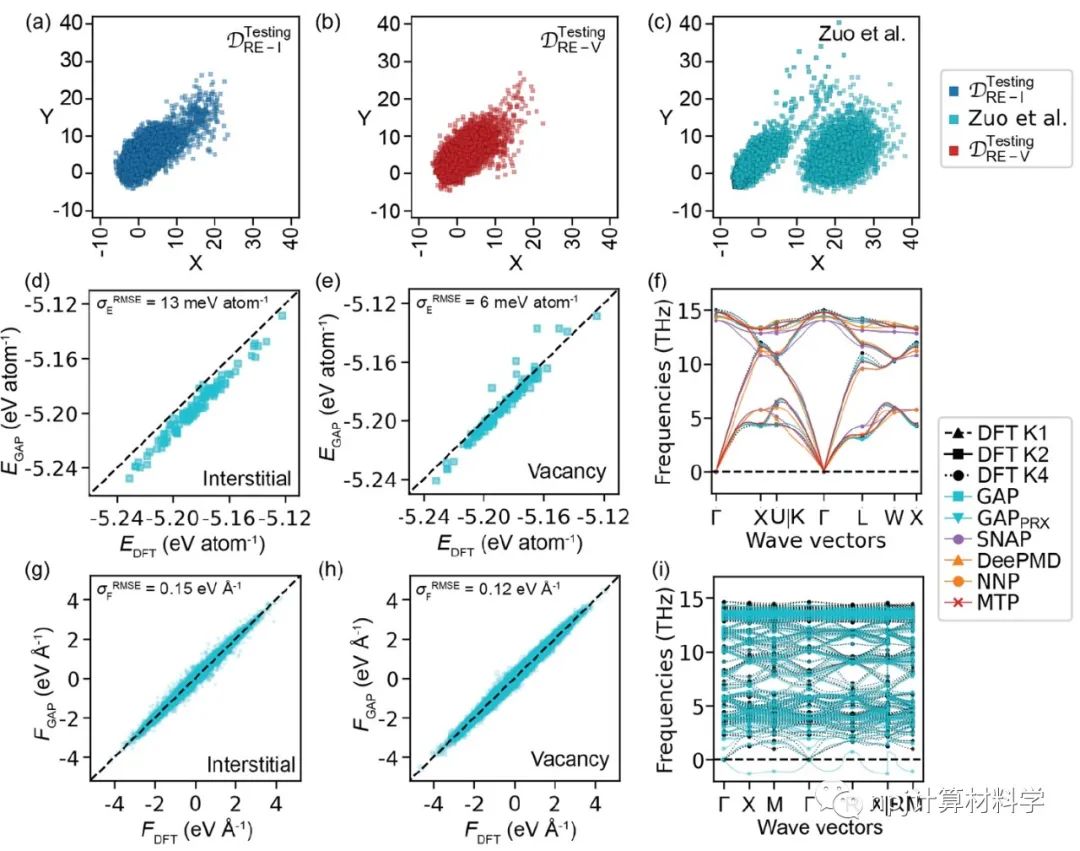
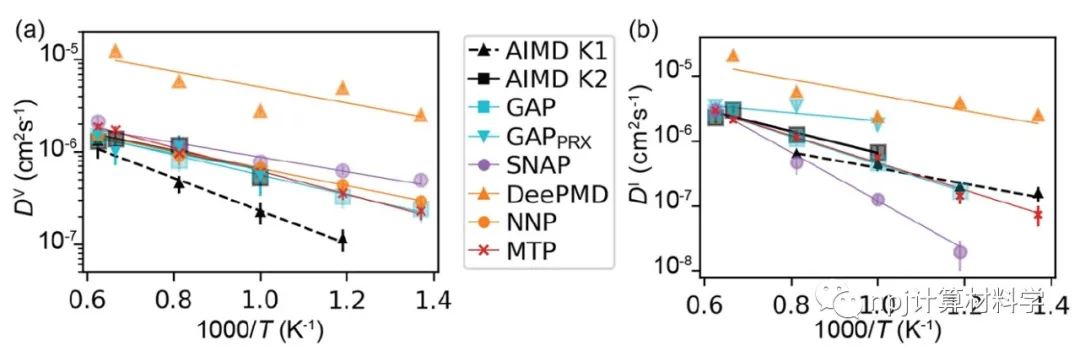
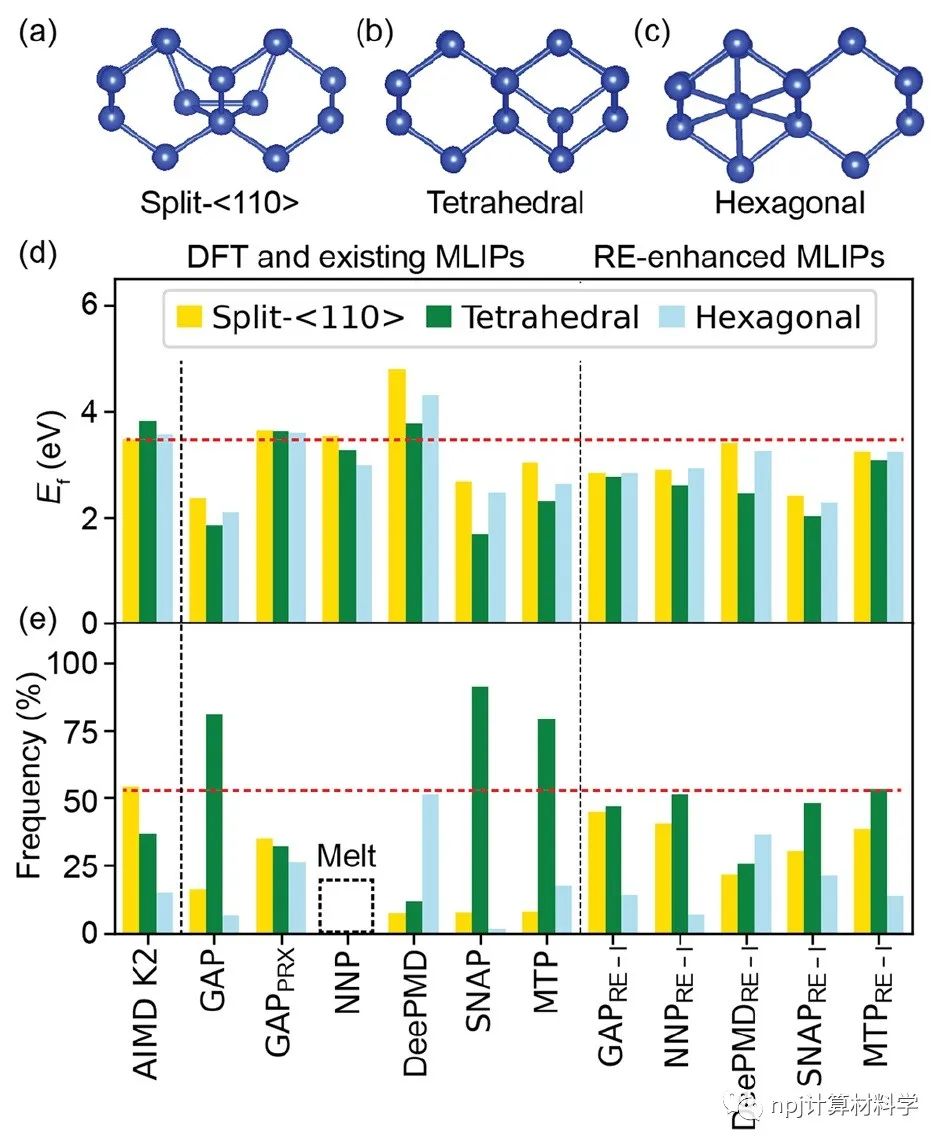
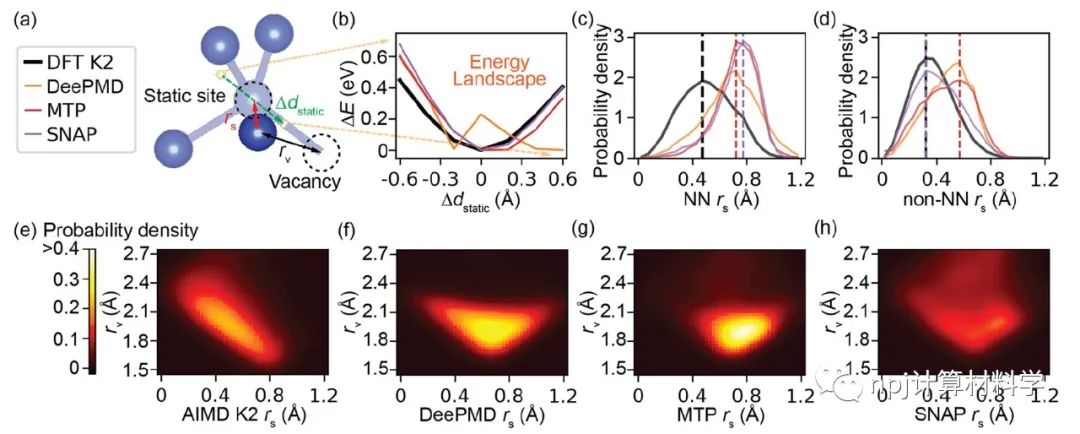
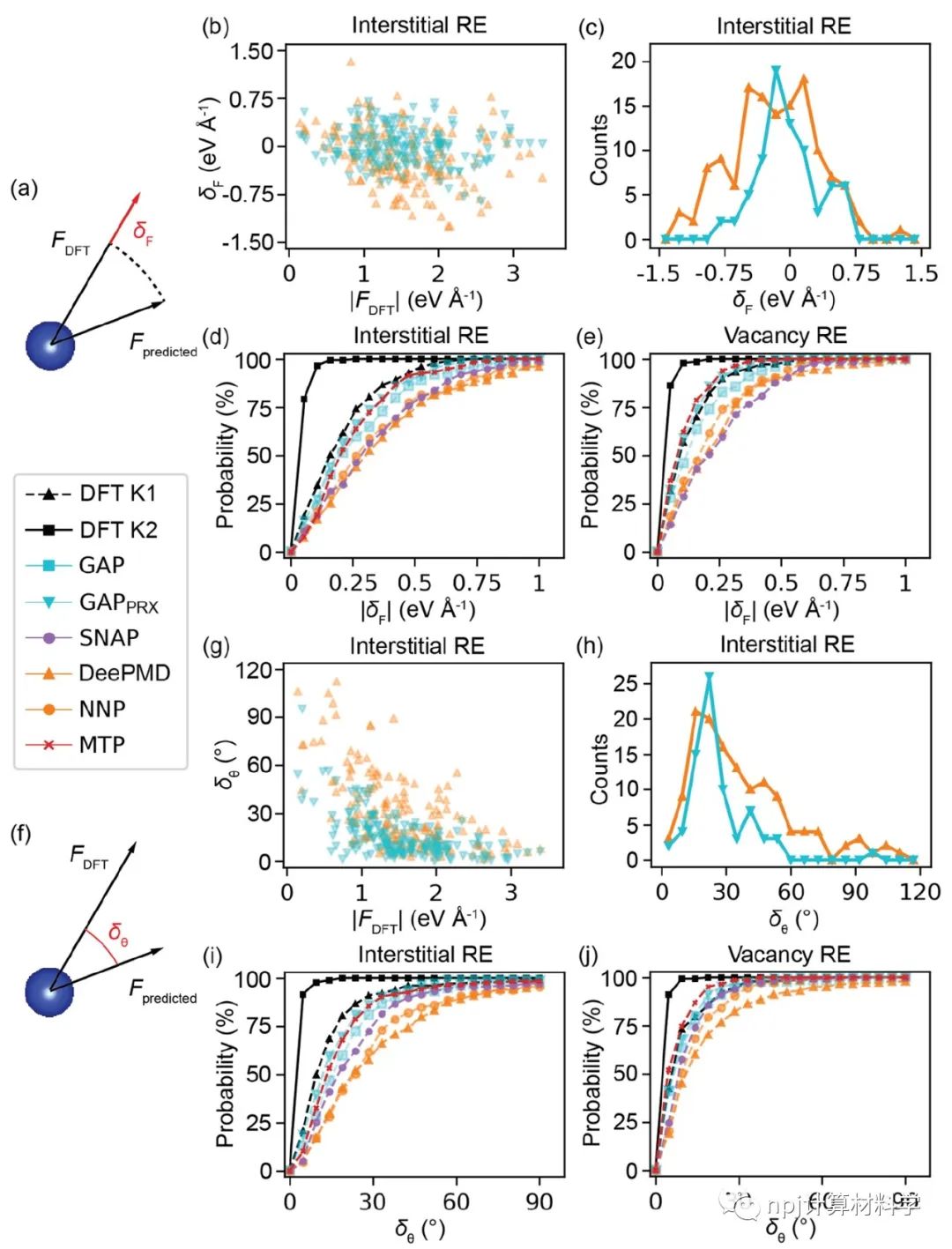
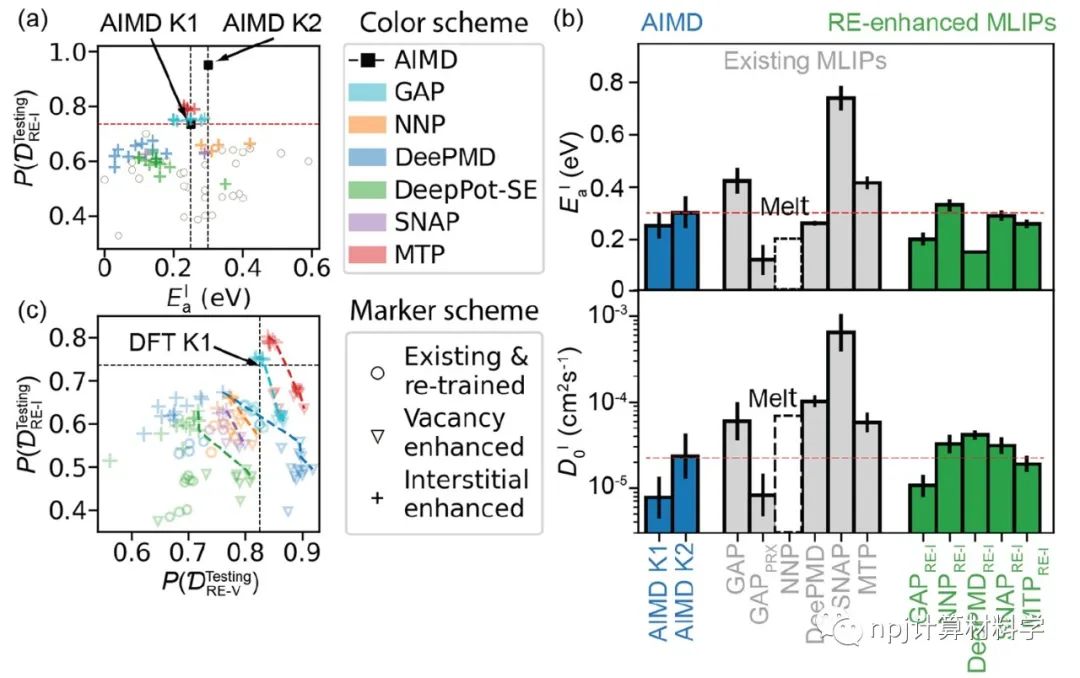
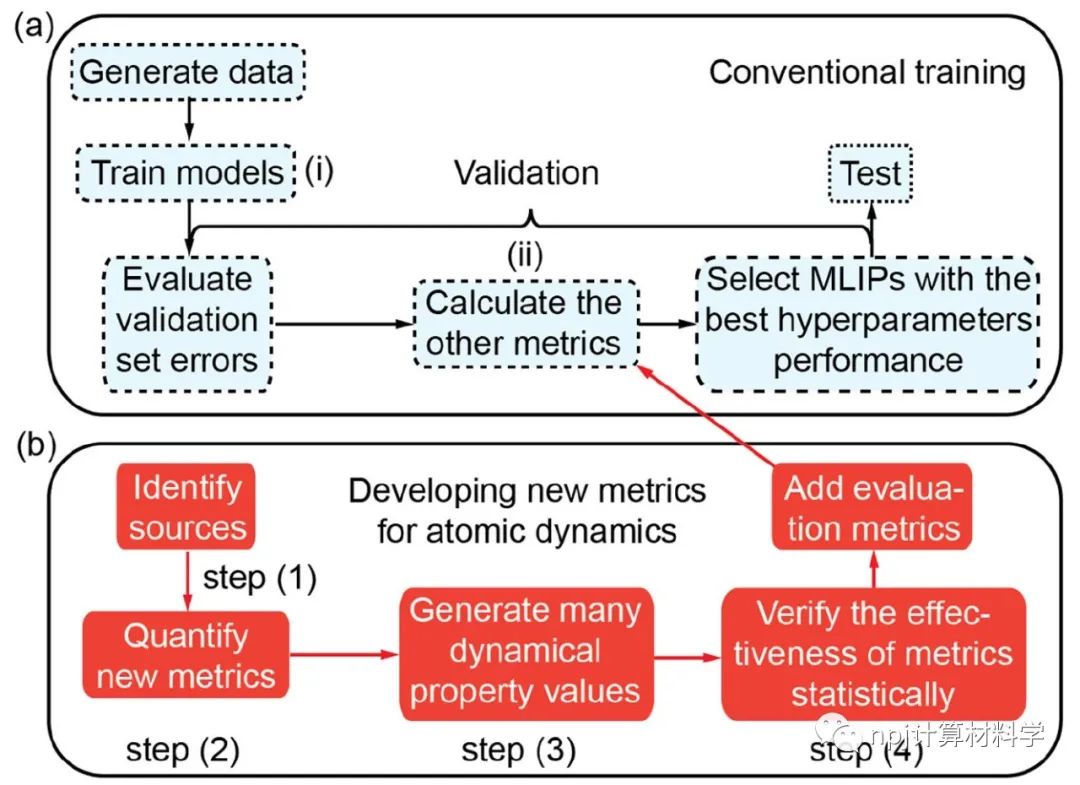
在原子模型领域,精确拟合原子间相互作用和计算量之间往往“鱼与熊掌不可兼得”:第一性原理计算长于拟合准确性却因计算量庞大而短于模拟大规模材料体系,经典原子势计算则恰恰相反。近来,机器学习原子势(machine learning interatomic potential,MLIP)作为一种新兴计算方法,极有潜力解决这一矛盾——在大规模模拟原子体系时保持接近第一性原理计算的准确性。Fig. 1 Testing of MLIPs.然而,尽管已有大量研究表明机器学习原子势能够精确拟合第一性原理计算(如密度泛函理论)所得的原子体系能量和原子作用力,其能否精确重现原子动力学现象和材料的物理性质这一问题始终悬而未决。Fig. 2 Diffusions of point defects in Si.来自美国马里兰大学材料科学和工程系的莫一非教授团队,通过系统性检测机器学习原子势和第一性原理计算在分子动力学模拟结果间的差异,识别出数个当下机器学习原子势的不足之处,提出了新的评价指标,并总结出一套有效的评价指标研究流程。Fig. 3 Si interstitials by MLIPs.作者们测试并总结了当前多种机器学习原子势在硅体系里的表现,观察到它们在分子动力学模拟中和第一性原理计算的原子运动方式有较大误差,并进而导致其预测材料物理性质时可能偏离原始值。机器学习原子势的不足之处集中在:1)原子运动(比如扩散和原子振动),2)缺陷和3)稀有事件这三方面。Fig. 4 Errors in atom vibrations.作者发现这些分子动力学模拟上的差异归咎于机器学习原子势在能量景观、形成能和在稀有事件原子上作用力的预测误差。他们随后针对稀有事件的原子作用力,提出全新的作用力表现分(force performance score)指标。该指标同时考虑作用力的大小误差和方向误差,能有效改进机器学习原子势的预测准确性。Fig. 5 Errors of atomic forces.他们将大量不同机器学习原子势的表现综合起来,展现了新评价指标和被预测的物理性质之间的显著关联。这一方法有助于评估指标的有效性并建立严谨的原子势检测流程。Fig. 6 The performance of RE-enhanced MLIPs.该研究揭示了传统的误差指标在评价机器学习原子势表现上的不足,为进一步改善机器学习原子势模型提供了严谨的数值依据和指导方向。作者提出的作用力表现分新评价指标流程可广泛应用于机器学习原子势的标准检测。相关论文近期发布于npj Computational Materials9: 174 (2023)。手机阅读原文,请点击本文底部左下角“阅读原文”,进入后亦可下载全文PDF文件。Fig. 7 Process of MLIP training and developing metrics.Editorial SummaryCitically assessing machine learning interatomic potentials’ performance.While first-principles computation, such as density functional theory (DFT), provides accurate description of atomic interactions in atomic modeling, its applications are limited to small materials systems of nanometer level and simulations lasting for a few nanoseconds. Alternatively, classical interatomic potentials offer large scale simulations of atomic systems, but they generally lack the accuracy as DFT calculations when describing interatomic bonds. Recently, machine learning interatomic potential (MLIP), as an emerging technique, shows a great opportunity to solve the dilemma between accuracies and computation cost in large-scale atomistic simulations. However, though many studies report that MLIPs have low errors fitting the energies and forces of atomic systems, whether MLIPs can accurately reproduce atomistic dynamics and physical properties of materials remains an open concern.A research team led by Prof. Yifei Mo from the Department of Materials Science and Engineering at the University of Maryland, USA, systematically investigated state-of-the-art MLIPs. Their comprehensive study revealed discrepancies between ab initio molecular dynamics (AIMD) and MLIP-MD simulations. They identified that these differences primarily manifest in atomic dynamics, including atomic vibrations, defects, and rare events. The discrepancies can be attributed to inaccurate predictions on energy landscapes, formation energies of defects, and forces on atoms involved in rare events. To address these, the team introduced novel evaluation metrics termed ‘force performance scores’ which consider both the force errors in magnitude and direction on rare-event atoms. By testing a number of MLIPs, they established correlations between the metrics and the physical properties predicted. This research not only emphasized the inadequacy of conventional error testing for evaluating MLIP performance but also provided robust insights and guidance to rectify the discrepancies observed in atomic dynamics, defects, and rare events. Their findings have been published in npj Computational Materials 9, 174 (2023).原文Abstract及其翻译Discrepancies and error evaluation metrics for machine learning interatomic potentials (机器学习原子势的误差及其评价指标)Yunsheng Liu, Xingfeng He & Yifei MoAbstract Machine learning interatomic potentials (MLIPs) are a promising technique for atomic modeling. While small errors are widely reported for MLIPs, an open concern is whether MLIPs can accurately reproduce atomistic dynamics and related physical properties in molecular dynamics (MD) simulations. In this study, we examine the state-of-the-art MLIPs and uncover several discrepancies related to atom dynamics, defects, and rare events (REs), compared to ab initio methods. We find that low averaged errors by current MLIP testing are insufficient and develop quantitative metrics that better indicate the accurate prediction of atomic dynamics by MLIPs. The MLIPs optimized by the RE-based evaluation metrics are demonstrated to have improved prediction in multiple properties. The identified errors, the evaluation metrics, and the proposed process of developing such metrics are general to MLIPs, thus providing valuable guidance for future testing and improvements of accurate and reliable MLIPs for atomistic modeling. 摘要机器学习原子势(MLIP)这一技术在原子建模领域极具潜力。大量的研究报告称机器学习原子势在数值拟合原子能量和力上能达到很小的误差。然而,机器学习原子势能否在分子动力学模拟中精确地复现原子层级的动力学现象和相关的物理性质却是这一技术未解的隐忧。本研究检验了当前数个机器学习原子势并发现它们和第一性原理计算的结果在原子动力学行为、缺陷和稀有事件上有相当的误差。该研究发现当下广泛用于测试机器学习原子势的平均能量误差和原子力误差等指标并不能充分地描述它们的表现。研究进而开发了其他量化指标,能更好地展现机器学习原子势在原子动力学方面的预测准确性。机器学习原子势在经过这类评价指标的优化后,在多个物理性质的预测上有显著的提高。本研究中所发现的误差,开发的评价指标和提出的开发流程可广泛应用于各类不同机器学习原子势,对于未来测试、改进、开发精确且可靠的机器学习原子势具有重要的指导意义。