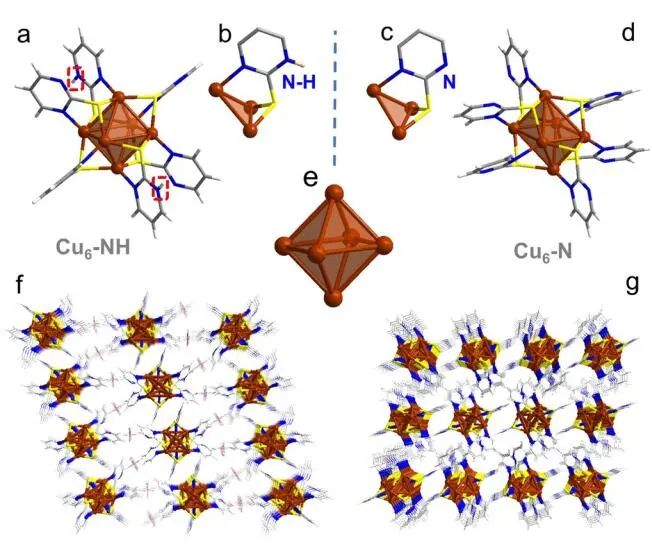
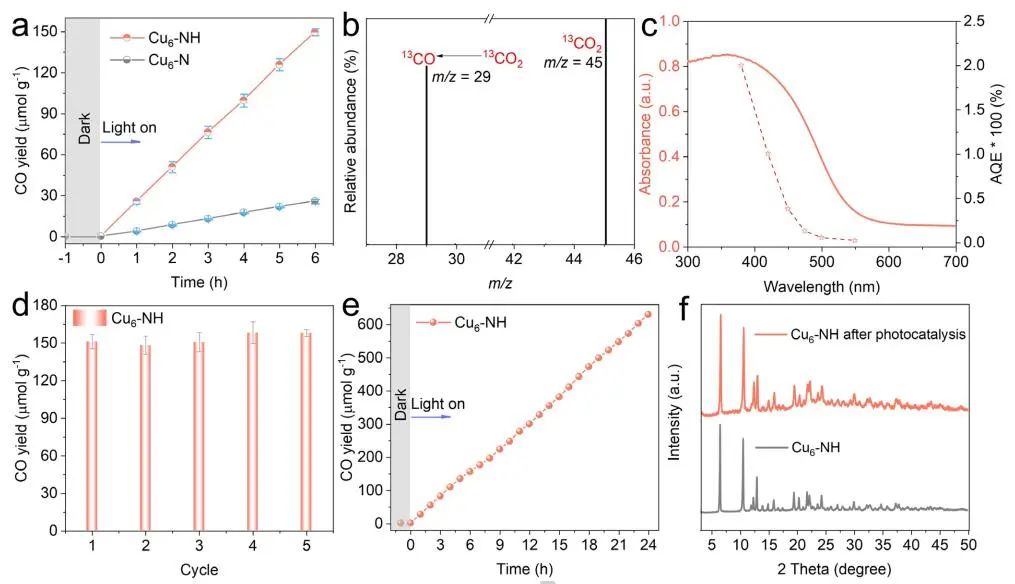
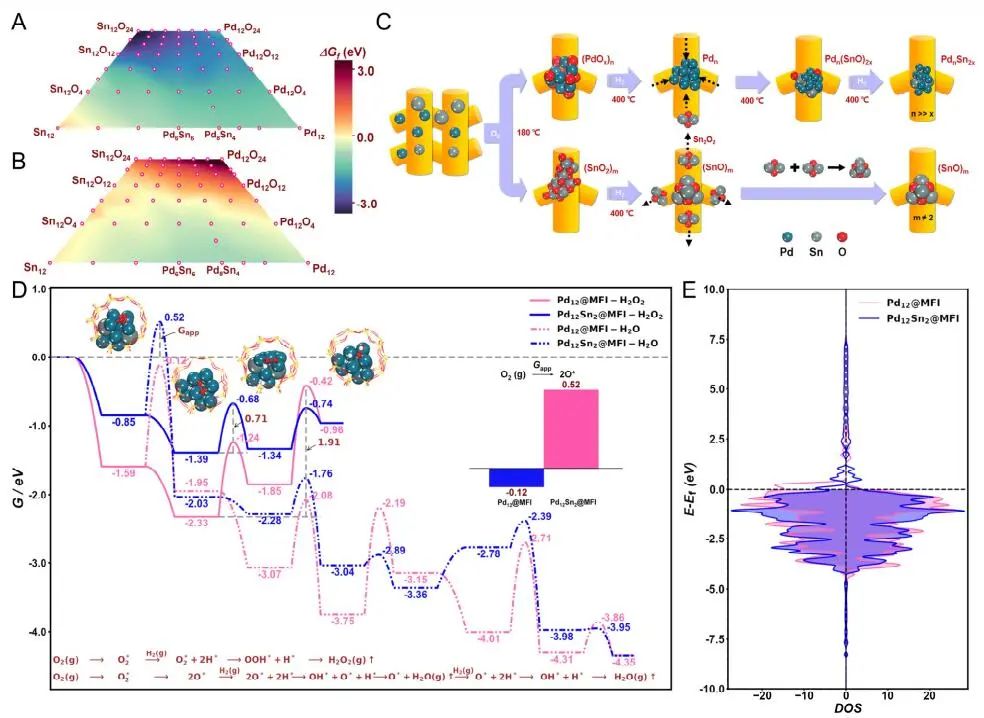
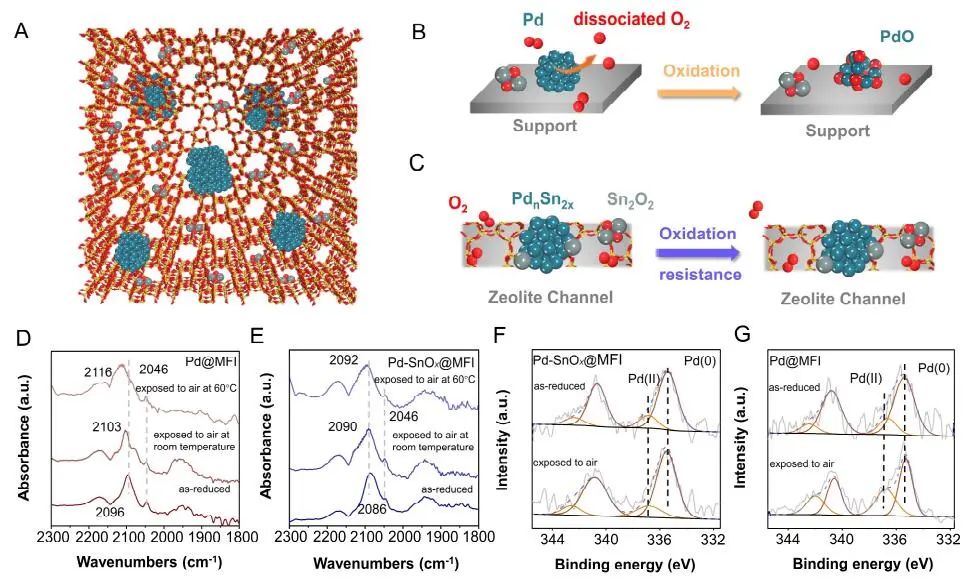
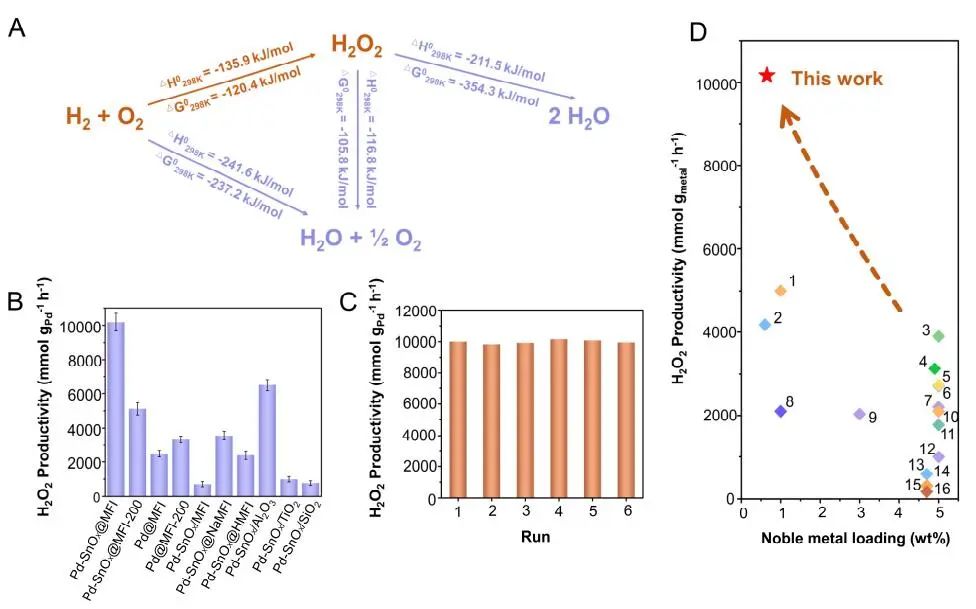
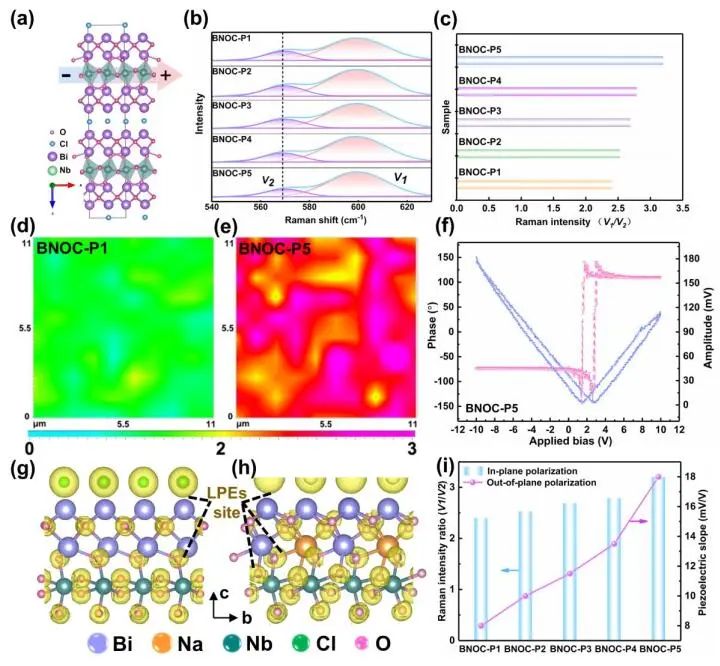
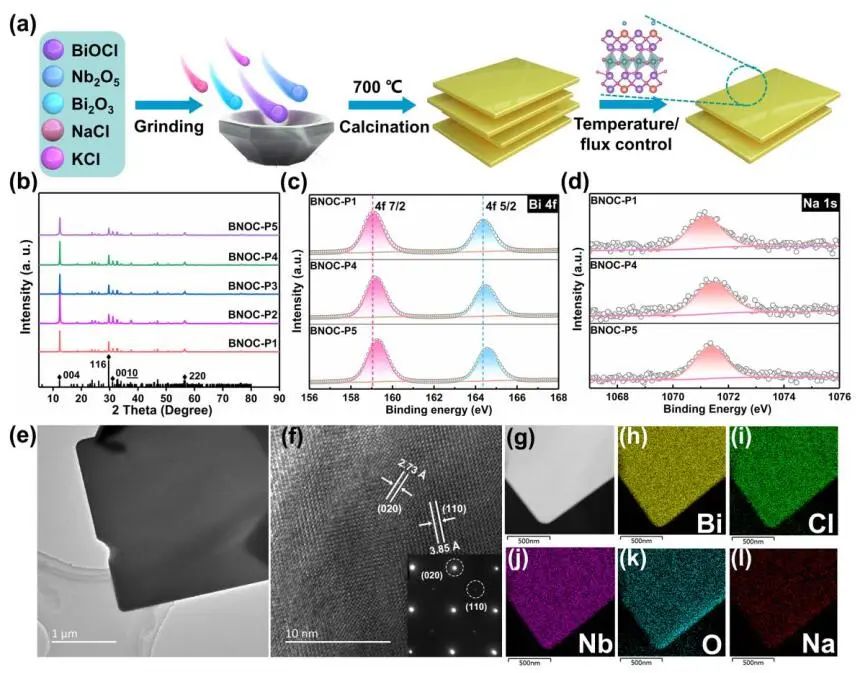
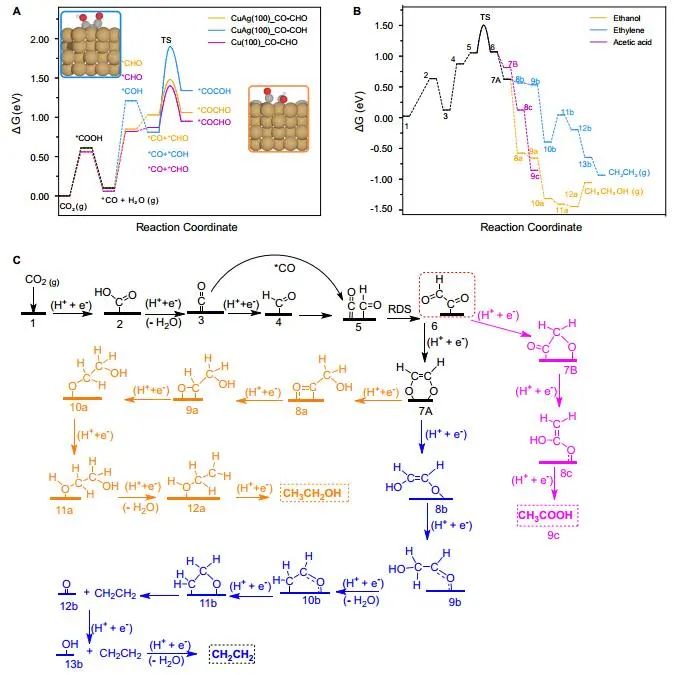
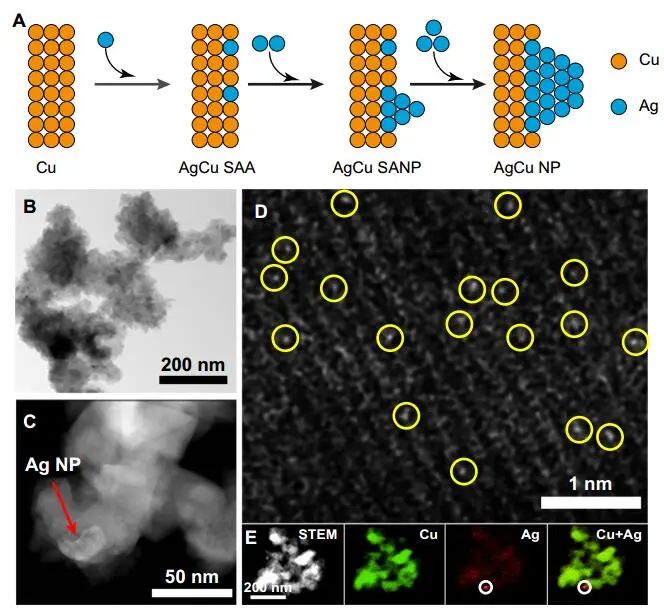
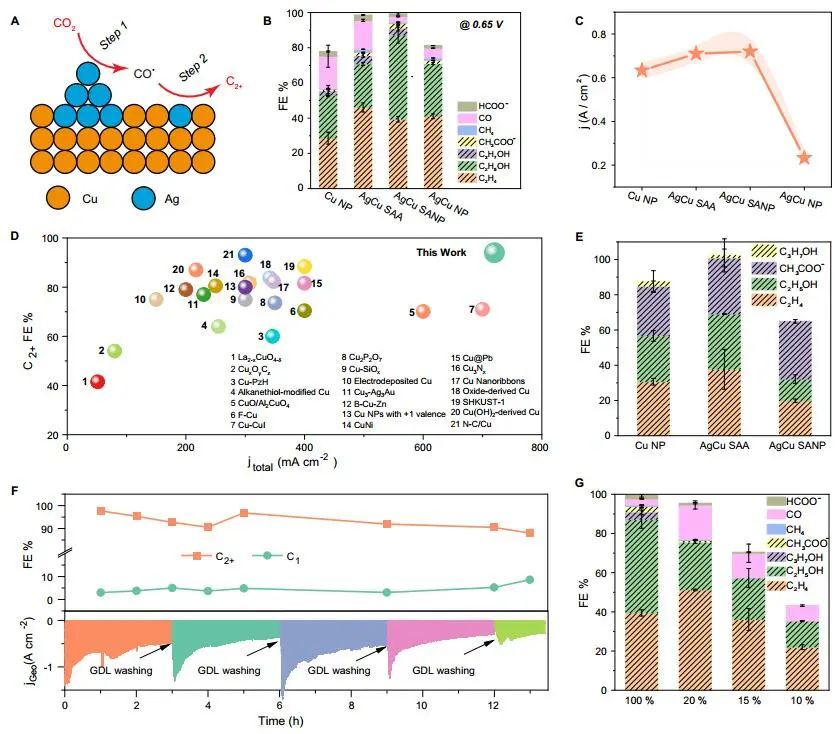
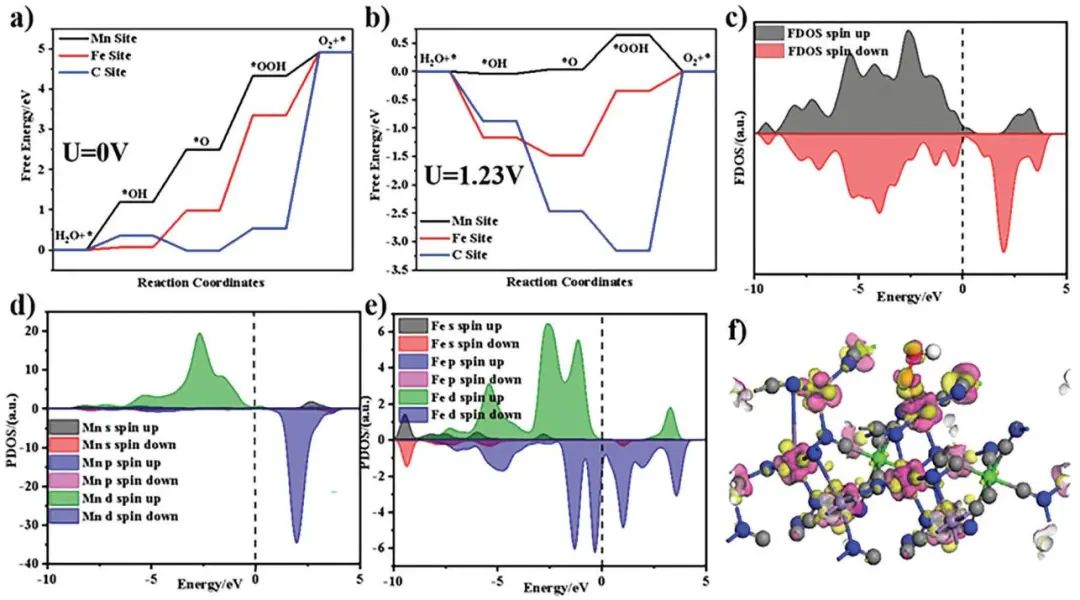
计算顶刊成果速递:3篇Angew、2篇Nat. Commun.、JACS、AFM、Small等! 2023年10月17日 下午2:52 • 顶刊 • 阅读 10 1.J. Am. Chem. Soc.:环境条件下ZnO上H2离解和COx加氢的原子尺度可视化 在原子尺度上研究氧化物表面的催化氢化反应一直具有挑战性,因为这些过程通常发生在环境或高压下,使得它们比原子尺度的技术更难实现。 基于此,上海科技大学杨帆副教授和中国科学技术大学李微雪教授等人报道了使用环境压力扫描隧道显微镜(AP-STM),环境压力X射线光电子能谱和密度泛函理论(DFT)计算对ZnO上H2解离和CO/CO2加氢的原子尺度研究。300 K时CO或CO2在ZnO表面的存在不会阻碍H2解离的表面位点的可用性,而CO甚至可以增强共吸附氢化物的稳定性,从而促进它们的解离吸附。 通过密度泛函理论(DFT)计算表明,CO2与氢化物在表面Zn3c位点上键合,可很容易地形成甲酸态。CO2与氢化物的结合能垒低至0.30 eV,反应能为0.09 eV。 形成的甲酸盐物种通过Zn-O键以单链构型(HCOOm*)结合在Zn3c位点上,再通过两个Zn-O键(HCOOb*)翻转与两个Zn3c位点形成双牙齿配位,从而发生结构弛缓,放热强度为-1.35 eV,势垒为0.21 eV。结果表明,CO2和H2在热力学和动力学上都有利于甲酸盐的生成。 DFT计算结果表明,首先CO与氢化物结合形成HCO*,吸热0.06 eV,能垒0.61 eV,然后形成的HCO*与表面晶格O进一步反应形成三齿构型(HCOOL*),能垒为1.06 eV,反应能为-0.39 eV。HCO*的势垒较低,更倾向于反向解离成CO和氢化物,因此CO很难直接氢化生成甲酸盐。 CO在ZnO(1010)上加氢为以下两步机制:CO首先在ZnO(1010)的⟨0001⟩步骤被氧化成CO2;生成的CO2在ZnO(1010)平台上扩散,与氢化物反应生成甲酸盐。 Atomic-Scale Visualization of Heterolytic H2 Dissociation and COx Hydrogenation on ZnO under Ambient Conditions. J. Am. Chem. Soc., 2023, DOI: https://doi.org/10.1021/jacs.3c08085. 2. Angew. Chem. Int. Ed.:首次报道!Cu-S-N簇助力CO2RR 原子精度高的铜(Cu)团簇是CO2还原反应(CO2RR)的理想催化剂,为阐述其结构-活性关系提供了合适的模型平台。然而,利用组装Cu团簇聚集体作为单组分光催化剂的高效全光催化CO2RR尚未见报道。 郑州大学臧双全教授等人报道了一种具有局部质子化N-H基团(记为Cu6-NH)的稳定晶体Cu-S-N簇光催化剂。该催化剂具有适宜的光催化氧化还原电位、高的结构稳定性、活性的催化物种和窄的带隙等特点,在可见光下具有出色的光催化CO2RR性能,CO的析出选择性近100%。 基于Cu6-NH和Cu6-N的单晶结构,Gibbs自由能计算发现,生成*CO2的Gibbs自由能低于初始值,说明CO2在Cu6-NH和Cu6-N上的吸附过程是放热的。计算结果表明,Cu6-NH和Cu6-N的*COOH的形成都是限速步骤。值得注意的是,Cu6-NH可以显著降低*COOH形成的能垒,从1.44 eV降低到0.79 eV,表明Cu6-NH中的质子中继站可以优化限速步骤,从而提高CO2光还原性能。 在CO2光还原过程中,Cu6-NH的能量消耗普遍低于Cu6-N,这是因为Cu6-NH具有高效的电荷转移特性,而质子化的嘧啶N作为质子中继站,可以显著降低*COOH中间体形成的能垒,是限速步骤。结果表明,Cu6-NH中的嘧啶质子确实可以参与CO2光还原,从而提供了一个低能的分子内加氢途径。 Copper-Sulfur-Nitrogen Cluster Providing a Local Proton for Efficient Carbon Dioxide Photoreduction. Angew. Chem. Int. Ed., 2023, DOI: https://doi.org/10.1002/anie.202313648. 3. Angew. Chem. Int. Ed.:Pd-SnOx@MFI高效催化合成过氧化氢 在空气中,金属表面容易因氧的O-O键解离而被氧化,从而降低其在各个领域的性能。虽然有几种配体修饰途径可减轻大块金属表面的氧化,但小尺寸金属纳米颗粒的抗氧化性还是一个挑战。 基于此,浙江大学王亮研究员等人报道了将小尺寸的Pd纳米颗粒固定在含锡(Sn)的MFI沸石晶体中(Pd-SnOx@MFI),Sn作为电子供体,通过削弱分子氧的吸附和抑制O-O裂解,有效地阻碍了Pd的氧化。Pd-SnOx@MFI在由氢气和氧气直接合成过氧化氢(H2O2)方面表现出优异的性能,H2O2的产率约为10170 mmol gPd-1 h-1,稳定性优于之前测试的催化剂。 由于沸石中同时存在Pd和Sn两种物种,少量的可移动Sn2O2有可能与Pdn发生碰撞形成PdnSn2O2。在还原条件下,由于PdxSny合金比纯Pdn或(SnO)n更稳定,生成的PdnSn2O2可被H2还原为PdnSn2合金。此外,两个Sn原子更倾向于原子分散在PdnSn2表面位置,这些位置暴露于直线或正弦通道或邻近的位置。因此,Pd-SnOx@MFI上的Sn可能由固定的PdxSny合金和捕获的(SnO)n组成。 对于沸石等氧化物载体外表面普遍负载的催化剂,从热力学角度考虑Pd-Sn合金是最稳定的,但键合效应限制了Sn物种在载体表面的移动,从而形成了阻碍Pt-Sn相互作用的动力学障碍,有利于形成相分离结构。 对于裸露的Pd表面,O2的吸附能为-1.59 eV,这种强烈的Pd-O2相互作用会导致Pd表面的O-O解理和氧化。Pd-d轨道的投影态密度(PDOS)表明,在金属Sn的存在下,Pd-d带的居群在费米能级附近降低。此外,Pd的d波段中心Pd12Sn2@MFI低于Pd12@MFI,表明Pd对O2的吸附减弱,O-O键解离受到抑制。 Efficient Catalytic Production of Hydrogen Peroxide Using Tin-containing Zeolite Fixed Palladium Nanoparticles with Oxidation Resistance. Angew. Chem. Int. Ed., 2023, DOI: https://doi.org/10.1002/anie.202312377. 4. Angew. Chem. Int. Ed.:Bi4NbO8Cl实现可见光驱动双功能水分解! 开发双功能水分解光催化剂具有重要的意义,但同时也面临着特殊晶面单晶的苛刻要求,这些单晶必须具有空间分离的活性位点和由良好的电荷驱动力所贡献的各向异性电荷转移路径。 基于此,中国地质大学(北京)黄洪伟教授等人报道了在Bi4NbO8Cl单晶纳米片中引入可调谐铁电极化,以增强正交电荷转移通道。测试发现,在牺牲试剂存在下,极性优化后的Bi4NbO8Cl在可见光照射下H2和O2的析出速率分别为54.21和36.08 μmol·h-1。 铁电磁滞回线显示,BNOC-P5比BNOC-P1和BNOC-P4表现出明显更高的剩余极化,说明其极化电场更强。对称振动模的强度对离子有效位移(Nb5+和O2-在八面体中)对拉曼激光诱导极化率的影响更为敏感,表明ν1和ν2的强度比可以反映NbO6八面体的畸变程度。 在BNOC-P5上观测到最大的强度比ν1/ν2,并且在拉曼图的扫描区域内,大的比值是均匀的,证明了BNOC-P5中NbO6八面体ab面内Nb5+阳离子强烈偏离中心。 此外,DFT计算的结构优化也揭示了NbO6八面体的畸变,这是由于Na+的引入缩小了[Bi2O2]2+层与钙钛矿层之间的层间间距。强烈的八面体畸变使BNOC纳米片具有较强的面内极化,有利于层内电荷的转移。 BNOC-P5的迟滞回线更陡,表明强的面外极化引发了电荷层间转移。因此,煅烧温度和磁通调制导致BNOC-P5的强极性铁电半导体同时具有较大的面内和面外极化。 Ferroelectric Polarization Modulated Facet-selective Charge Separation in Bi4NbO8Cl Single Crystal for Boosting Visible-light Driven Bifunctional Water Splitting. Angew. Chem. Int. Ed., 2023, DOI: https://doi.org/10.1002/anie.202312895. 5. Adv. Funct. Mater.:NiCo2O4/NF在高电流密度下电催化GOR! 电催化甘油氧化反应(GOR)是将生物质副产品转化为高附加值化学品的有效途径,但现有催化剂的氧化活性和转化率较低。基于此,中国科学院上海硅酸盐研究所施剑林院士和崔香枝研究员等人报道了通过Ni取代Co3O4中的八面体Co3+,可控的制备了NiCo2O4/NF双金属氧化物纳米阵列,其在高电流密度(E300=1.42 V,E600=1.62 V)下具有优异的GOR催化活性,在1.42 V下具有97.5%的总法拉第效率(FE)(FEformic acid=89.9%和FEglycolic acid=7.62%)。GOR/析氢耦合双电极电解槽电压比50 mA cm−2下的水分解电压低约299 mV,同时该电解槽在1.75 V下稳定运行200 h以上,能耗降低16.9%,在阳极获得高附加值产品。 通过DFT计算,作者研究了Co3O4/NF和NiCo2O4/NF对GOR的潜在活性来源。Co3O4/NF对甘油(-0.984 eV)和OH*(0.415 eV)的吸附能分别增加到NiCo2O4/NF的-1.151(CoOh位点)和0.756 eV(NiOh位点),表明Ni的引入可以优化甘油和OH*的共吸附性能。 在整个反应过程中,Co3O4/NF的速率决定步骤(RDS)是C3H6O3*转化为C3H5O3*(步骤6→7),绝对速率阶能为1.38 eV,而NiCo2O4/NF的RDS为CH2O2*解吸(步骤19→20),绝对速率阶能为1.00 eV。与Co3O4/NF相比,Ni的引入促进了GOR热力学。结果表明,在Co3O4/NF中引入Ni可显著优化甘油和OH*的共吸附行为,降低了RDS能垒,从而实现了优异的GOR性能。 Controllable Electron Distribution Reconstruction of Spinel NiCo2O4 Boosting Glycerol Oxidation at Elevated Current Density. Adv. Funct. Mater., 2023, DOI: https://doi.org/10.1002/adfm.202306995. 6. Nat. Commun.:揭示缺陷碳基电催化剂的动态活性位点 无金属碳材料中活性位点的确定对于开发实用的电催化剂至关重要,但由于反应过程中动态结构演变过程难以捉摸,因此确定活性位点的精确构型还是一个挑战。 基于此,吉林大学姚向东教授和澳大利亚卧龙岗大学陈俊等人报道了通过对缺陷密度和碳上O-基团类型的可控操纵来揭示缺陷位点的性质,包括构建缺陷密度与过氧化氢(H2O2)法拉第效率之间的相关性。结果表明,缺陷密度最高的氧修饰缺陷石墨烯-30(O-DG-30)催化剂具有优异的H2O2法拉第效率(~98.38±1.6%)。 通过DFT计算,作者研究了有关活性羰基修饰五边形缺陷性质的原子见解。鉴于醌和羰基在O-DG-30上共存,作者提出了多种模型结构,主要包括在缺陷部位的锯齿形边缘和扶手椅形边缘上锚定的潜在O-基所建立的构型。 对于2e− ORR,热力学平衡势对应于活火山图的顶点。在理论上,具有最大活性的催化剂应有4.22 eV的GOOH*。C1和C2模型的ORR活性最低,说明羧基不是影响O-DG-30 ORR活性的主要因素。 D2模型活性最高,UL和ΔG OOH*值分别为0.683 V和4.217 eV,非常接近理论理想值,B1和A1模型在火山地块上的位置与D2模型非常接近。 需注意,O基团锚定在位于锯齿形和扶手椅边交界处的六边形上,而不是碳晶格中的富集位点。羰基修饰的五边形缺陷(C5=O)可视为O-DG-30生成H2O2的主要活性位点,因此O-DG-30催化剂具有最高的缺陷密度,为特定的O基团修饰提供了足够的位点,从而最大化了2e– ORR性能。 Unveiling the dynamic active site of defective carbon-based electrocatalysts for hydrogen peroxide production. Nat. Commun., 2023, DOI: https://doi.org/10.1038/s41467-023-41947-7. 7. Nat. Commun.:AgCu单原子合金和Ag纳米颗粒级联电催化CO2还原 电催化CO2还原成增值多碳(C2+)产物提供了一种利用可再生电力关闭人为碳循环的手段,但C2+产物的催化选择性不理想严重阻碍了该技术的实际应用。 基于此,加拿大滑铁卢大学吴一民教授和卡尔加里大学Samira Siahrostami等人报道了一个级联的AgCu单原子合金和纳米颗粒电催化剂,其中Ag纳米颗粒产生CO,AgCu单原子合金促进C-C耦合动力学。结果表明,在所制备的AgCu单原子合金和纳米颗粒催化剂下,在-0.65 V、~720 mA cm-2工作条件下,对C2+产物的法拉第效率(FE)为94±4%。 通过DFT计算,作者研究了在Ag掺杂的Cu催化剂表面上对乙烯、乙醇和乙酸进行原子尺度的CO2还原反应机理。CO2RR到C2+产物涉及多种中间体,其中C-C偶联是决定速率的步骤。Cu(100)上*CO加氢成*CHO和*COH的势垒分别为0.64和0.94 eV,比CO-CO直接偶联更有利。因此,作者选择研究Ag掺杂Cu(100)表面上能量较低的C-C耦合势垒,即*CO-*CHO和*CO-*COH。 作者还计算了*CO-*CHO在纯Cu(100)表面的TS势垒,为0.55 eV,比Ag掺杂Cu(100)表面高0.1 eV。Ag位点在单原子合金(AgCu SAA)和纳米颗粒(AgCu SANP)中均能将CO2转化为CO,但后者由于活性位较多,预计会促进CO的形成。 结果表明,在CO2还原到CO方面,Ag掺杂Cu是比纯Ag表面更好的催化剂。低配位的Ag原子对CO2转化为CO的反应最活跃,AgCu SANP催化剂预计具有扭结和角点。因此,CO2到CO的还原很可能同时发生在单个Ag原子和Ag纳米颗粒上。 Cascade electrocatalysis via AgCu single-atom alloy and Ag nanoparticles in CO2 electroreduction toward multicarbon products. Nat. Commun., 2023, DOI: https://doi.org/10.1038/s41467-023-41871-w. 8. Small:MnFe-PBA@IF助力室温下OER和超级电容器! 提高析氧反应(OER)速率的关键在于加速四-电子动力学,而促进超级电容器发展的关键在于电极材料的设计。基于此,青岛大学唐建国教授和姜倩倩副教授、武汉工程大学黄华波副教授等人报道了在室温下合成了锰铁普鲁士蓝(MnFe-PBA@IF),并采用快速还原基质制备了六边形凹形结构。根据MnFe-PBA的吉布斯自由能计算,Mn原子的电荷耗尽可以大大增强表面富电子含氧基团的吸附。1 M KOH的过电位为280 mV,同时它还可以作为超级电容器,稳定的工作电压范围为-0.9至0 V,比容量为1260 F g−1。 通过DFT计算,作者对MnFe-PBA的活性表面和电子结构进行了深入分析。在原始MnFe-PBA(101)表面模型上,作者选择了三个活性位点Mn、Fe和C。N位点(3.04)和O位点(3.44)具有很强的电负性,因此氮原子不能与含氧中间体吸附。即使将含氧中间体放在N的顶部,经过几何优化后,它仍然会被反弹。三个MnFe-PBA位点的都吸附和解吸*OOH、*OH和*O,从而加速中间产物的转化。 此外,Mn、Fe和C活性位点的最大上坡能分别为1.835、2.369和4.388 eV,因此三个活性位点的理论过电位分别为0.605、1.139和3.158 V。在这种情况下,原始MnFe-PBA(101)板下的速率决定步骤来自于*O到*OOH的转化,即最大的上坡步骤是由*OOH的形成引起的。 由于Mn的电负性低于Fe的电负性,Mn向Fe的部分牵引和转移发生,因此Mn与吸附的含氧中间体的结合中等强度,可能是由于更好的OER活性。Mn和Fe与吸附的含氧中间体的结合较弱,导致产生较大的过电位,最明显的是*OOH中间体,同时最突出的C位点是*中间体。 Unlocking the Potential of Amorphous Prussian Blue with Highly Active Mn Sites at Room Temperature for Impressive Oxygen Evolution Reaction and Super Capacitor Electrochemical Performance. Small, 2023, DOI: https://doi.org/10.1002/smll.202303946. 原创文章,作者:v-suan,如若转载,请注明来源华算科技,注明出处:https://www.v-suan.com/index.php/2023/10/17/09f9672343/ 赞 (0) 0 0 生成海报 相关推荐 Nature子刊:原位监测:铜(I)氧化物上两步氧化形成Zhang-Rice单态触发水氧化 2023年10月11日 宁平&张秋林ACS Catalysis:受阻路易斯酸碱对提高Ni/CeO2纳米催化剂的低温CO2甲烷化性能 2023年10月26日 JACS:调制Au@Pd纳米线上Pd 4d轨道重叠程度,实现选择性制备H2O2 2024年2月5日 北科刘永畅,最新JACS!钠电领域进展 2023年10月16日 北化工汪乐余/殷雄Adv. Sci.:制备亲水SACs一般策略!助力高效选择性加氢 2023年11月15日 周豪慎/乔羽AM:通过构建电极正面定制阳离子的溶剂化鞘层 2023年10月14日 发表回复 请登录后评论...登录后才能评论 提交